
Editor’s note: Today we hear from Atommap, a computational drug discovery company that has built an elastic supercomputing cluster on the Google Cloud to empower large-scale, computation-driven drug discovery. Read on to learn more.
Bringing a new medicine to patients typically happens in four stages: (1) target identification that selects the protein target associated with the disease, (2) molecular discovery that finds the new molecule modulating the function of the target, (3) clinical trial that tests the candidate drug molecule’s safety and efficacy in patients, and (4) commercialization that distributes the drug to patients in needs. The molecular discovery stage, in which novel drug molecules are invented, involves solving two problems: first, we need to establish an effective mechanism to modulate the target function that maximizes the therapeutic efficacy and minimizes the adverse effect; second, we need to design, select, and make the right drug molecule that faithfully implements the mechanism, is bioavailable, and has acceptable toxicity.
What makes molecular discovery hard?
A protein is in constant thermal motion, which changes its shape (conformation) and binding partners (other biomolecules), thus affecting its functions. Structural detail of a protein’s conformational dynamics time and again suggests novel mechanisms of functional modulation. But such information often eludes experimental determination, despite tremendous progress in experimental techniques in recent years.
The chemical “universe” of all possible distinct small molecules — estimated to number 1060 (Reymond et al. 2010) — is vast. Chemists have made probably ten billion so far, so we still have about 1060 to go.
There lie two major challenges of molecular discovery and its endless opportunity: chances are that we have not considered all the mechanisms of action or found the best molecules, thus we can always invent a better drug.
Atommap’s computation-driven approach to molecular discovery
Harnessing the power of high-performance computing, Atommap’s molecular engineering platform enables the discovery of novel drug molecules against previously intractable targets through new mechanisms, making the process faster, cheaper, and more likely to succeed. In past projects, Atommap’s platform has dramatically reduced both the time (by more than half) and cost (by 80%) of molecular discovery. For example, it played a pivotal role in advancing a molecule against a challenging therapeutic target to the clinical trial in 17 months (NCT04609579) and it substantially accelerated the discovery of novel molecules that degrade high-valued oncological targets (Mostofian et al. 2023).
Atommap achieves this by:
-
Advanced molecular dynamics (MD) simulations that unveil complex conformational dynamics of the protein target and its interactions with the drug molecules and other biomolecules. They establish the dynamics-function relationship for the target protein, which is instrumental to choosing the best mechanism of action for the drug molecules.
-
Generative models that enumerate novel molecules. Beginning with a three-dimensional blueprint of a drug molecule’s interaction with its target, our models computationally generate thousands to hundreds of thousands of new virtual molecules, which are designed to form the desired interactions and to satisfy both synthetic feasibility and favorable drug-like properties.
-
Physics-based, ML-enhanced predictive models that accurately predict molecular potencies and other properties. Every molecular design is evaluated computationally for its target-binding affinity, its effects on the target, and its drug-likeness. This allows us to explore many times more molecules than can be synthesized and tested in the wet lab, and to perform multiple rounds of designs while waiting for often-lengthy experimental evaluation, leading to compressed timelines and increased probability of success.
Computation as a Service and Molecular Discovery as a Service
To truly, broadly impact drug discovery, Atommap needs to augment its deep expertise in molecular discovery by partnering with external expertise in the other stages — target identification, clinical trials, and commercialization. We form partnerships in two ways: Computation as a Service (CaaS) and Molecular Discovery as a Service (MDaaS, pronounced Midas), which make it easy and economically attractive for every drug discovery organization to access our computation-driven molecular engineering platform.
Instead of selling software subscriptions, Atommap’s pay-as-you-go CaaS model lets any discovery project first try our computational tools at a small and affordable scale, without committing too much budget. Not every project is amenable to computational solutions, but most are. This approach allows every drug discovery project to introduce the appropriate computations cheaply and quickly, with demonstrable impact, and then deploy them at scale to amplify their benefits.
For drug hunters who would like to convert their biological and clinical hypotheses into drug candidates, our MDaaS partnership allows them to quickly identify potent molecules with novel intellectual property for clinical trials. Atommap executes the molecular discovery project from the first molecule (initial hits) to the last molecule (development candidates), freeing our partners to focus on biological and clinical validation.
The need for elastic computing
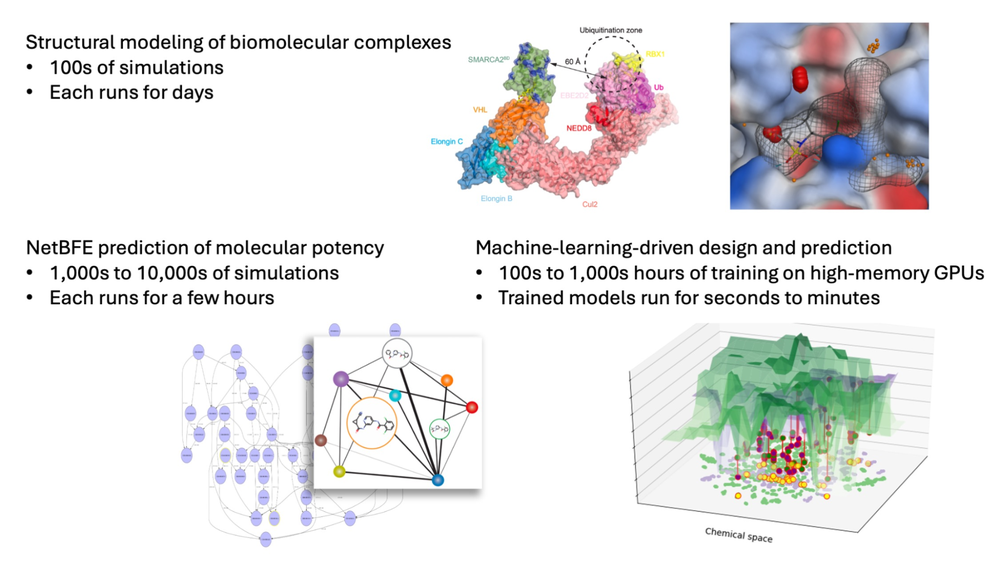
Figure 1. Diverse computational tasks in Atommap’s molecular engineering platform require elastic computing resources.
For Atommap, the number of partnership projects and the scale of computation in each project fluctuate over time. In building structural models to enable structure-based drug design, we run hundreds of long-timescale MD simulations on high-performance GPUs to explore the conformational ensembles of proteins and complexes between proteins and small molecules, each of which can last hours to days. Our NetBFE platform for predicting the binding affinities invokes thousands, sometimes tens of thousands, of MD simulations, although each one is relatively short and completes in a few hours. Atommap’s machine learning (ML) models take days to weeks to train on high-memory GPUs, but once trained and deployed in a project, run in seconds to minutes. Balancing the different computational loads associated with different applications poses a challenge to the computing infrastructure.
To meet this elastic demand, we chose to supplement our internal computer clusters with Google Cloud.
How to build an elastic supercomputer on Google Cloud
It took us several steps to move our computing platform from our internal cluster to a hybrid environment that includes Google Cloud.
Slurm
Many workflows in our platform depended on Slurm for managing the computing jobs. To migrate to Google Cloud, we built a cloud-based Slurm cluster using Cloud HPC Toolkit, an open-source utility developed by Google. Cloud HPC Toolkit is a command line tool that makes it easy to stand up connected and secure cloud HPC systems. With this Slurm cluster up and running in minutes, we quickly put it to use with our Slurm-native tooling to set up computing jobs for our discovery projects.
Cloud HPC Toolkit naturally fits our DevOps function into best practices. We defined our compute clusters as “blueprints” within YAML files that allow us to simply and transparently configure specific details of individual Google Cloud products. The Toolkit transpiles blueprints into input scripts that are executed with Hashicorp’s Terraform, an industry standard tool for defining “infrastructure-as-code” such that it can be committed, reviewed, and version-controlled. Within the blueprint we also defined our compute machine image through a startup script that’s compatible with Hashicorp’s Packer. This allowed us to easily “bake in” the software our jobs typically need, such as conda, Docker, and Docker container images that provide dependencies such as AMBER, OpenMM, and PyTorch.
The deployed Slurm cloud system is as accessible and user-friendly as any Slurm system we have used before. The compute nodes are not deployed until requested and are spun down when finished, thus we only pay for what we use; the only persistent nodes are the head and controller nodes that we log into and deploy from.
Batch
Compared to Slurm, the cloud-native Google Batch gives us even greater flexibility in accessing the computing resources. Batch is a managed cloud job-scheduling service, meaning it can be used to schedule cloud resources for long-running scientific computing jobs. Virtual machines that Batch spins up can easily mount either NFS stores or Google Cloud Storage buckets, the latter of which are particularly suitable for holding our multi-gigabyte MD trajectories and thus useful as output directories for our long-running simulations.
Running our workflows on Google Cloud through the Batch involves two steps: 1) copying the input files to Google Cloud storage, 2) submitting the batch job.
- code_block
- <ListValue: [StructValue([('code', '$ gcloud storage cp -R ./local_input_dir gs://my-gcs-bucket/work_dirrn$ gcloud batch jobs submit example-job –config=./job_cfg.json –location=us-central1'), ('language', ''), ('caption', <wagtail.rich_text.RichText object at 0x3e1508d181f0>)])]>
SURF
A common pattern has emerged in most of our computational workflows. First, each job has a set of complex input files, including the sequence and structures of the target protein, a list of small molecules and their valence and three-dimensional structures, and the simulation and model parameters. Second, most computing jobs take hours to days to finish even on the highest-performance machines. Third, the computing jobs produce output datasets of substantial volume and subject to a variety of analyses.
Accordingly, we have recently developed a new computing infrastructure, SURF (submit, upload, run, fetch), which seamlessly integrates our internal cluster and Google Cloud through one simple interface and automatically brings the data to where it is needed by computation or the computation to where the data resides.
- code_block
- <ListValue: [StructValue([('code', '$ am-surf submit my_job/ –num-cpus 16 –num-gpus 8 –where [gcp|internal]'), ('language', ''), ('caption', <wagtail.rich_text.RichText object at 0x3e1508d18dc0>)])]>
SURF submits jobs to Google Cloud Batch using Google’s Python API.
We now have an elastic supercomputer on the cloud that gives us massive computing power when we need it. It empowers us to explore the vast chemical space at an unprecedented scale and to invent molecules that better human health and life.
Andrew Sabol supported us from the very beginning of Atommap, even before we knew whether we could afford the computing bills. Without the guidance and technical support of Vincent Beltrani, Mike Sabol, and other Google colleagues, we could not have rebuilt our computing platform on Google Cloud in such a short time. Our discovery partners put their trust in our young company and our burgeoning platform; their collaborations helped us validate our platform in real discovery projects and substantially improve its throughput, robustness, and predictive accuracy.